

Парацетамол: терапевтическое применение и проблема острых отравлений
Е. М. ШИФМАН1, А. Л. ЕРШОВ2
1 Республиканский перинатальный центр, Петрозаводск, Республика Карелия
2 Всероссийский центр экстренной и радиационной медицины МЧС России, Санкт-Петербург
Статья опубликована в журнале ОБЩАЯ РЕАНИМАТОЛОГИЯ, 2007, III; 1
В форме обзора литературы обсуждаются вопросы механизмов действия и терапевтического применения одного из самых известных и широко применяемых лекарственных средств - парацетамол. В 1995 г. эксперты ВОЗ провели сравнительную оценку препаратов разных групп с сочетанным анальгетическим и антипиретическим действием. По критерию «эффективность/безопасность» первое место получил парацетамол. Интерес к механизму действия и обсуждение безопасности применения возобновились с новой силой в связи с появившейся возможностью применения внутривенной формы парацетамола для обезболивания в послеоперационном периоде. На основании обзора литературы, авторы делают вывод о том, что парацетамол может с успехом использоваться в практической работе ОРИТ. Этот препарат обладает собственным достаточно выраженным обезболивающим и антипиретическим эффектом, а его совместное использование с наркотическими анальгетиками приводит к синергизму действия и снижению расхода опиоидов. При применении в терапевтических дозах парацетамол практически безопасен, но требует взвешенного подхода при назначении пациентам из групп риска.
Как это нередко бывает в медицине, история открытия парацетамола (П) связана со случайным стечением обстоя тельств. В 1893 г. по ошибке фармацевта больному с хроническим болевым синдромом в состав лекарства попало постороннее химическое соединение — ацетанилид. К счастью, пациент не только не пострадал, но и отметил выраженное уменьшение боли. Дальнейшие исследования ацетанилида показали, что это вещество является достаточно мощным анальгетиком, но даже в умеренных терапевтических дозах приводит к тяжелому токсическому повреждению печени [1]. В дальнейшем были предприняты энергичные попытки синтезировать новый анальгетик, основанный на химической структуре ацетанилида, но обладающий меньшей токсичностью. Вскоре на свет появился препарат, названный в Англии «парацетамол» (от пара-ацетил-амино-фенол), а в США - «ацетаминофен» [1]. По химической структуре П представляет собой n-ацетаминофенол: С8H9NO2 с молекулярной массой 151,17 и температурой плавления около 170°С. Парацетамол почти нерастворим в воде, но хорошо растворим в спирте и ацетоне, а также в едких щелочах. Структурная формула П отображена на рис.1.
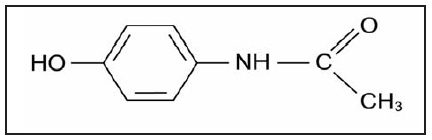 |
| Рис.1. Структура парацетамола |
Настоящее признание и широчайшая популярность П наступили через полвека — после того, как в 1949 г. в Великобритании была разработана технология промышленного производства высокоочищенного препарата [2]. Началу массового производства П способствовал высокий спрос населения на лекарственные средства, одновременно обладающие выраженным противоболевым и жаропонижающим действием при минимуме побочных эффектов. Как оказалось, П хорошо соответствует этим требованиям. В 1995 г. эксперты ВОЗ провели сравнительную оценку препаратов разных групп с сочетанным анальгетическим и антипиретическим действием. По критерию «эффективность/безопасность» первое место получил П [1]. Среди анальгетиков- антипиретиков П продолжает оставаться лидером по продажам даже спустя 100 лет от момента своего первого клинического применения и почти через 60 лет после широкого выхода на потребительский рынок [1].
В настоящее время П выпускается в разных странах почти под 200 названиями. В числе наиболее известных в России синонимов П для перорального приема можно упомянуть Ацетаминофен, Панадол, Тиленол, Калпол. Кроме того, П входит в состав многих комбинированных препаратов, таких как Анаколд, Залдиар, Иралгезик, Каффетин, Ринзасип, Гриппостад С, Седал-М и др. Существуют также формы препарата для внутривенного введения, но пока объем их выпуска сравнительно невелик. На отечественный рынок поставляется раствор П под названием «Перфалган». До недавнего времени в клиниках западных стран для внутривенного введения использовался «пропрепарат» П под торговой маркой «Пропацетамол».
Некоторые сведения о возможных механизмах терапевтического действия парацетамола
Несмотря на длительную историю клинического применения и хорошую изученность терапевтических эффектов, механизм действия П до настоящего времени остается не вполне
понятным.
К началу 70-х гг. ХХ в. не вызывало сомнений, что П обладает преимущественно центральным механизмом действия
[3—5] и то, что анальгетический эффект П до определенной
степени пропорционален как разовой, так и суточной (суммарной) дозе препарата [6].
Определенный прорыв в понимании механизмов действия
ненаркотических анальгетиков наступил около 35 лет назад,
когда было показано, что большинство нестероидных противовоспалительных препаратов (НСПВП) реализует терапевтическое действие за счет подавления синтеза простагландинов. Как
оказалось, ключевую роль играет воздействие НСПВП на фермент циклооксигеназу (ЦОГ или простагландин-Н2-синтетаза)
[7]. Десятью годами позже данное открытие принесло Джону
Вэйну (John Vane) Нобелевскую премию по медицине.
Еще через 10 лет стало очевидным, что существуют по
крайней мере две изоформы ЦОГ, названные соответственно
ЦОГ-1 и ЦОГ-2 [8]. Большинство известных в настоящее время НСПВП одновременно блокирует ЦОГ-1 и ЦОГ-2 [9].
Имеется точка зрения, поддерживающая наличие селективности препаратов в отношении этих ферментов. Она, безусловно,
имеет право на существование, но пока еще нуждается в тщательно построенных доказательствах. Некоторые исследования, выполненные в самые последние годы, позволяют допустить существование еще одного представителя семейства ЦОГ
- ЦОГ-3 [10, 11].
В настоящее время считается доказанным, что ЦОГ-1 активно участвует как в воспалительных процессах, так и в механизмах возникновения боли. В эксперименте было продемонстрировано возрастание активности ЦОГ-1 в клетках глии
спинного мозга во время болевой стимуляции. В то же время, интратекальное введение препарата «SC-560», способного избирательно ингибировать ЦОГ-1, приводило к существенному снижению ощущения боли у экспериментальных животных [12].
ЦОГ-2 также является активным участником воспалительного процесса. Применение специфических ингибиторов
указанного фермента сопровождается заметным противовоспалительным и анальгетическим эффектом. Мы считаем, что в
данном случае термин «специфичность», как и «селективность», еще ждет свои критерии истины, доказывающие их
право на признание. Интересно, что опухолевые клетки при
некоторых вариантах рака экспрессируют рецепторы к ЦОГ-2,
что делает потенциально возможным создание новых противоопухолевых препаратов на основе ЦОГ-2 [2].
Оказалось, П — очень слабый ингибитор как ЦОГ-1, так и
ЦОГ-2, хотя его клиническое применение сопровождается достаточно выраженным анальгетическим и антипиретическим
эффектом по сравнению с другими препаратами [1, 2]. Это обстоятельство внесло дополнительные сложности в понимание
механизмов действия П, но позволило объяснить практически
полное отсутствие у препарата противовоспалительного эффекта (противовоспалительное действие многих препаратов
реализуется через ингибирование ЦОГ-2).
В самые последние годы было высказано два предположения о механизмах действия П. Graham et al. [13] соглашаются,
что препарат практически не оказывает влияния in vitro на ферменты ЦОГ-1 и ЦОГ-2, но считают, что П способен блокировать
in vivo биологические эффекты простагландинов в интактных
клетках за счет прямого уменьшения концентрации арахидоновой кислоты. Возможно, влияние П на синтез простагландинов
опосредуется через регуляцию стимулирующих воздействий на
этот процесс провоспалительных цитокинов [13].
По мнению других исследователей, П является специфическим и единственным ингибитором гипотетической ЦОГ-3
[2]. Предполагается также, что роль ЦОГ-3 реализуется на
сравнительно поздних стадиях воспалительного процесса и
опосредуется через влияние на синтез эндогенных противовоспалительных медиаторов [14, 15]. Действие П может также
быть связано с центральными механизмами, отличными от ингибирования синтеза простагландинов.
Возможности применения парацетамола для обезболивания в послеоперационном периоде
В медицинских исследованиях нередко в качестве «золотого стандарта» обезболивания в послеоперационном периоде выступают опиоиды [16, 17]. Наркотические анальгетики обладают мощным действием при болях различного генеза, интенсивности и локализации, а также позитивно изменяют эмоциональный фон прооперированного больного. Однако применение препаратов этой группы потенциально опасно из-за большого числа побочных эффектов, среди которых наиболее очевидные — угнетение дыхания, задержка мочи, ослабление перистальтики кишечника, рвота и т. д. Вероятность развития патологической зависимости от наркотических анальгетиков у больного также является ограничивающим фактором их клинического применения. Существуют различные методики, направленные на минимизацию применения наркотических анальгетиков при сохранении высокого качества послеоперационного обезболивания. Так, в 1992—1993 гг. Kehlet и Dahl предложили свой подход к решению данной задачи [18, 19]. Они указали, что одновременное назначение больному в послеоперационном периоде анальгетиков с различным механизмом действия способствует не только снижению доз вводимых препаратов, но и уменьшению частоты возникновения побочных эффектов, свойственных каждому компоненту в комбинации лекарств. В частности, такой эффект был отмечен при следующих сочетаниях: «наркотический анальгетик + НСПВП» [20], а также «наркотический анальгетик + П» [21—23].
В своем недавнем аналитическом обзоре Remy [24] отмечает, что первый из указанных вариантов комбинированной терапии не лишен недостатков. В их число вошла невозможность реализации указанного подхода приблизительно у 25% прооперированных больных из-за тех или иных противопоказаний к применению НСПВП [25]. Кроме того, у больных, получивших в послеоперационном периоде смесь наркотического анальгетика и НСПВП, нередко наблюдались существенные изменения в свертывающей системе крови, которые заставляли либо ограничить применение указанной методики в послеоперационном периоде, либо полностью от нее отказаться из- за ухудшения гемостаза [26].
В отличие от НСПВП, П имеет сравнительно небольшое количество противопоказаний к применению и в терапевтических дозах редко вызывает клинически значимые побочные эффекты [27]. Кроме того, П может быть назначен в послеоперационном периоде пациентам любого возраста. Эти качества П привлекательны для анестезиологов-реаниматологов и способствуют его применению как в комбинации с другими анальгетиками, так и в варианте монотерапии.
В ряде работ последнего десятилетия был показан синергизм обезболивающего действия препаратов при одновременном назначении П и опиатов [21—23], что позволяло статистически достоверно сокращать дозу вводимых наркотиков [28]. У пациентов с различной хирургической патологией было наглядно продемонстрировано снижение потребности в назначении опиатов в первые сутки послеоперационного периода [23, 29—33].
Как указывалось выше, число и тяжесть осложнений при применении опиатов зависит от вводимой дозы препарата. Исходя из этого, можно было ожидать, что комбинация наркотических анальгетиков с П будет способствовать уменьшению числа негативных эффектов, связанных с применением опиатов. Однако результаты дальнейших исследований оказались не столь однозначными. При послеоперационном обезболивании комбинацией морфина и П у лиц, перенесших операции на конечностях, сердце и позвоночнике, частота возникновения рвоты статистически не отличалась от таковой в контрольной группе больных, получавших только морфин [30, 32, 34]. Также между сопоставляемыми группами не отмечалось достоверных различий по глубине седативного эффекта [30, 32, 33] и частоте задержки мочи [32, 35]. В то же время, большинство исследователей указывают на достоверное уменьшение в послеоперационном периоде частоты депрессии дыхания при одновременном применении П с морфином по сравнению с группой пациентов, которым вводился морфин без П [23, 29, 30].
Только в работе Sinatra et al. не подтверждается достоверное снижение частоты возникновения угнетения дыхания при одновременном введении опиатов и П [35]. В некоторых исследованиях сопоставлялись результаты опросов больных, касающихся уровня их удовлетворенности послеоперационным обезболиванием [30, 31, 35]. Ни в одном случае не было отмечено существенных различий между группами больных, получавших только морфин и морфин в комбинации с П. Таким образом, применение сочетанного обезболивания (опиоид + П) в послеоперационном периоде позволяет снизить необходимую дозу наркотических препаратов и, возможно, вероятность развития депрессии дыхания, но не способно существенно повлиять на вероятность развития тошноты и рвоты, задержки мочи, чрезмерной седации больных. Надо отметить, что снижение потребности в морфине было наиболее отчетливым у больных, имеющих болевой синдром умеренной степени выраженности (среднее потребление морфина снизилось на 37%), и менее заметным у пациентов с ярко выраженной болью (среднее снижение потребности в морфине составило 18%) [24].
Подробное рассмотрение возможностей применения в послеоперационном периоде сочетания «морфин + НСПВП» лежит за рамками тематики настоящего обзора. Тем не менее, сопоставление опубликованных результатов использования комбинаций «морфин + П» и «морфин + НСПВП» показало лучший обезболивающий эффект у последнего сочетания [29]. В то же время, при изолированном (без морфина) использовании в постоперационном периоде, обезболивающий эффект П оказался выше, чем у НСПВП [5].
Некоторые возможные механизмы реализации гепатотоксичного действия парацетамола
Как указывалось выше, в терапевтических дозировках П обладает сравнительно небольшой токсичностью, однако осознанный или случайный прием высоких доз препарата способен приводить к тяжелому поражению печени по типу центролобулярного гепатонекроза, нередко заканчивающегося смертью пациента [36]. Возможно, что по этой причине именно П уже достаточно долгое время является наиболее популярным лекарственным препаратом для отравлений в суицидных целях в США [37, 38]. Среди больных, поступивших в последнее десятилетие в один из стационаров США в связи с острым отравлением П, 86% пришлось на суицидные попытки и лишь 14% — на случайные передозировки препарата [39]. Однако, согласно статистике, представленной одним из специализированных центров по лечению острых отравлений США, число лиц, госпитализи рованных в связи со случайными отравлениями П, составило 65%, а на долю суицидальных отравлений пришлось 35% [38]. В Великобритании среди госпитализированных пациентов с острыми отравлениями П 84% пришлось на суицидные попытки [40]. Очень близкие к этим цифрам результаты были получены и в Дании (85% от госпитализированных — результат суицидных попыток) [41, 42].
В последние годы в зарубежной медицинской периодике было опубликовано несколько интересных обзоров литературы, обобщающих последние данные по гепатотоксичности П [43—46]. Первоначальные биохимические и метаболические эффекты, возникающие в организме пострадавшего в острейшей стадии отравления, хорошо изучены. Но патологические процессы, ведущие к повреждению печеночных клеток при передозировке П, остаются во многом неразгаданными. С высокой степенью определенности можно утверждать, что в этом случае речь должна идти о варианте гибели клеток, не связанном с апоптозом [47, 48].
Дополнительный свет на проблему гепатотоксичности П пролили работы, выполненные в разные годы в лаборатории, возглавляемой доктором Gillette. Около 30 лет назад было показано, что обмен П в организме человека происходит при участии цитохрома Р450. В результате прямой двухэлектронной оксидации образуется активный метаболит П — N-ацетил-р- бензохинонимин (NAPQI), способный к ковалентной связи с белком [49]. Около 10 лет назад было показано, что способностью к превращению в организме П в активный метаболит NAPQI (путем оксидации) также обладают цитохромы 2Е1, 1A2, 3A4 и 2А6 [50-52]. Далее, в лаборатории д-ра Gillette было продемонстрировано, что в процессах детоксикации NAPQI принимает участие глутатион (GSH), образующий конъюгаты с П. При введении подопытным животным токсических доз П происходит снижение уровня GSH в печени почти на 90%, что обусловлено формированием ковалентных связей между цистеином, входящим в состав глутатиона, и NAPQI. Таким образом, формируются П-белковые стабильные радикалы (аддукты), которые обладают невысокой токсичностью и могут быть элиминированы из организма [49]. В схематическом виде процессы формирования токсических метаболитов П и пути их элиминации из организма представлены на рис.2.
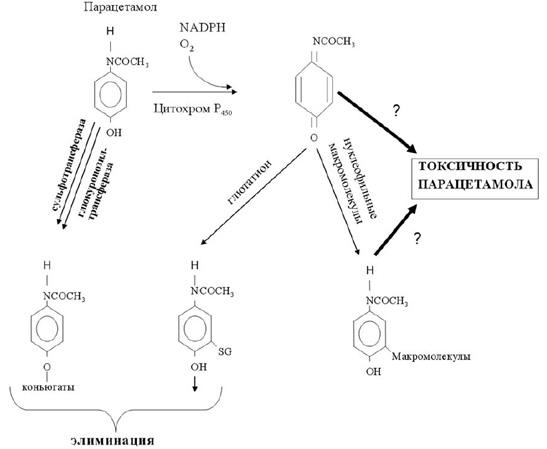 |
| Рис.2. Обмен парацетамола в организме, формирование токсических метаболитов, процессы детоксикации и элиминации (по: James et al., 2003 [44, 45]). |
Комплекс «парацетамол-белок».
В опубликованных за последние 10-15 лет работах было
показано, что при отравлении П тяжесть общего состояния
экспериментальных животных хорошо коррелирует с концентрацией комплекса «парацетамол-белок» («П-белок»), для
оценки которого в крови используется методика ELISA [53,
54]. Установлено, что химическая связь между П и белком носит ковалентный характер и при формировании комплекса
«П-белок» задействована аминокислота цистеин [16]. Сравнительно недавно лабораторная методика определения концентрации комплекса «П-белок» была внедрена в клиническую
практику. Положительные результаты анализа позволяют
уточнить парацетамоловую этиологию острой печеночной недостаточности и верифицировать факт отравления П [55].
Вероятно, комплекс «П-белок» является не только лабораторным маркером отравления П, но и выступает в качестве одного из важнейших патогенетических факторов, приводящих к поражению гепатоцитов. Пока получен ограниченный
объем информации о механизмах воздействия комплекса «П-
белок» на клетки печени. Предполагается, что возникновение
ковалентной связи между П и наиболее биологически значимыми внутриклеточными белками печени сопровождается
следующей цепью событий: снижение активности гепатоцитов, затем — гибель и лизис клеток [44, 45]. Согласно этой гипотезе белком-мишенью для П являются митохондриальные
протеины клеток печени. Ковалентное соединение препарата с
внутриклеточными белками сопровождается снижением энергетических процессов в гепатоците и, как следствие, нарушениями в трансмембранном ионном градиенте [46, 56], а также
резким угнетением активности плазматической АТФазы [57].
К настоящему времени идентифицировано около 20 белков, способных образовывать ковалентные связи с П. Среди них глутаминсинтетаза, глутаминдегидрогеназа, альдегиддегидрогеназа, глутатионпероксидаза, карбоангидраза III, глутаматдегидрогеназа, глицин-N-метилтрансфераза и др. [58]. Есть основания предполагать, что ингибирование большинства внутриклеточных ферментов гепатоцитов не носит немедленный и тотальный характер. В исследованиях James et al. [44, 45] было показано, что через 2 часа после введения экспериментальным животным токсической дозы П активность глутаматдегидрогеназы и N-10-формилтетрагидрофолатдегидрогеназы в гепатоцитах снизилась приблизительно на 25%. В то же время, частичное, но совпадающее во времени, снижение активности значительного числа внутриклеточных энзимов может оказаться достаточным для необратимых нарушений функций клетки и ее гибели. Требуются новые исследования, позволяющие детализировать роль повреждения внутриклеточных ферментативных механизмов в формировании острой печеночной недостаточности при отравлениях П.
Оксидативный стресс.
Оксидативный стресс также является одним из возможных механизмов реализации токсических эффектов высоких
доз П. Повышенное образование супероксида по механизму
реакции Фентона ведет к образованию перекиси водорода и гидроксильного радикала (•ОН). (В реакцию Фентона (Men+ + Н2О2>Me(n+1)+ + •OH + OH- или Men+ + ROOН > Me(n+1)+ + RO• + H2O) вовлекаются катионы металлов переходной валентности в низшей степени окисления (Fe2+, Cu+, Ti3+, Cr2+, Co2+). Высокая эффективность продукции активных форм кислорода в реакции Фентона обеспечивается сопряженной реакцией, в которой супероксид-анион-радикал выступает в роли восстановителя: Me(n+1)+ + •О2- > Men+ + О2.) Было продемонстрировано,
что скорость реакции между NAPQI и GSH очень высока (k1 =
3,2 x 104 М-1s-1 при значении рН = 7,0) [59].
Как указывалось выше, после приема токсических доз П в
высоких концентрациях образуется NAPQI, что, как следствие, сопровождается выраженным уменьшением уровня GSH
в центролобулярных клетках печени. Предполагается, что в
подобной ситуации происходит ингибирование GSH-пероксидазы — наиболее важного энзима, принимающего участие в
процессах детоксикации. Кроме того, в процессе образования NAPQI под влиянием цитохрома Р450 происходит образование супероксид-анионов (•ОО-), обладающих мощным цито
токсическим действием. Под влиянием супероксиддисмутазы
супероксид-анионы превращаются в кислород и перекись водорода [60].
Накапливается все больше данных, указывающих на высокую вовлеченность оксидативного стресса в гепатотоксичность П. Nakae et al. [61] продемонстрировали в опытах на крысах резкое возрастание токсических свойств препарата при одновременном назначении инкапсулированной супероксиддисмутазы. Кроме того, на экспериментальных животных было показано, что дефероксамин (комплексообразующий препарат, используемый при лечении отравлений препаратами железа) способен ослабить токсические свойства высоких доз П [62]. Несколько позже выяснилось, что дефероксамин не столько предотвращает токсическое действие П на печень, сколько задерживает развитие этого повреждения (в среднем на 24 часа) [63]. Все перечисленные данные позволяют предполагать: в реализации оксидативного стресса и повреждении клеток печени при отравлении П может играть активную роль реакция Хабера-Вейсса, катализатором в которой выступает железо [44, 45, 64].
Роль купферовских клеток.
В нескольких лабораториях западных стран проводят постоянную работу по изучению роли макрофагальных клеток в
токсичности П [65]. В печени макрофагальными клетками являются клетки Купфера. Известно, что в процессе жизнедеятельности указанных клеток продуцируется ряд биологически
активных молекул, среди которых гидролитические ферменты, эйкозаноиды, оксид азота, супероксид. Кроме них, в ходе
контакта с П, купферовские клетки способны высвобождать
некоторые провоспалительные цитокины, в число которых
входят Il-1, Il-6 и TNF-? [66—69].
Laskin et al. [65] изучали in vivo механизмы токсического
воздействия П на купферовские клетки. Им была предложена
экспериментальная модель, в которой физиологическая активность купферовских клеток искусственно угнеталась за счет
введения хлорида гадолина и сульфата декстрана. Выяснилось,
что крысы, которым предварительно вводили эти вещества, существенно лучше переносили токсические дозы П. Схожие результаты были получены и на мышах [66]. Goldin et al. [70] показали, что блокада купферовских клеток липосомами,
содержащими дихлорметилена дифосфанат, также достоверно
снижает токсический эффект высоких доз П. Полученные данные позволили авторам высказать предположение о существенной роли клеток Купфера в механизмах развития отравления П.
Однако в недавней работе Ju et al. [71] были получены не
столь однозначные результаты. Действительно, введение дихлорметилена дифосфаната позволяло эффективно снижать
токсичность. В то же время выяснилось, что при введении хлорида гадолина наступает ингибирование физиологической активности лишь у части купферовских клеток, что ставило под
сомнение трактовку полученных ранее результатов. Таким образом, роль клеток Купфера в процессе реализации токсических эффектов П требует дальнейшего изучения.
Нитротирозин и токсичность парацетамола.
Сравнительно недавно в опытах на экспериментальных
животных было обнаружено, что введение токсических доз П
сопровождается накоплением нитротирозина в клетках печени,
расположенных центролобулярно [72]. Данные иммуногистохимического анализа свидетельствуют о том, что нитраты образуются именно в тех клетках, в которых происходит накопление аддукторов П, а позднее развивается некроз. В организме
млекопитающих пероксинитрит представляет собой одно из
немногих химических соединений, способных превращать тирозин в нитротирозин. Есть основания предполагать, что именно это соединение принимает участие в метаболизме П в клетках печени, приводя к накоплению продукта реакции —
нитротирозина. Вместе с тем известно, что пероксинитрит —
весьма агрессивный оксидант, потенциально способный повреждать многие клеточные структуры печени [73]. В условиях
ослабления антиоксидантной защиты повреждающие эффекты
пероксинитрита становятся еще более значимыми [74]. Анион
пероксинитрита является сравнительно стабильным, хотя в условиях пониженного рН период его полураспада уменьшается,
составляя около 1,9 секунды. Реакции оксидации, вызываемые
пероксинитритом, могут протекать с участием одного или двух
электронов. Доказана роль этого соединения в процессах оксидации липидов, белков и ДНК [73].
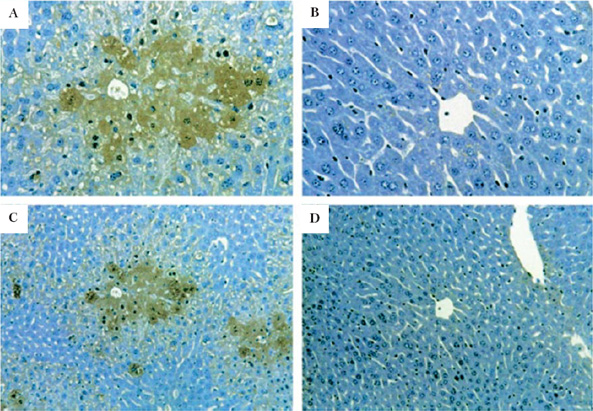 |
| Рис. 3. Иммуногистохимическое определение нитротирозина в клетках печени в экспериментальной модели острого отравления парацетамолом на мышах. А: мыши введено П 300 мг/кг, животное забито через 4 часа после начала опыта (х40), бурые пятна отражают накопления клетками нитротирозина. В: печень мыши, которой был введен только физиологический раствор. С и D: те же гистологические препараты (х20) (цит. по: James L. et al., 2003 [44], с разрешения авторов). |
Таким образом, процесс оксидации клеточных структур, провоцируемый высокими дозами П и реализуемый при участии пероксинитрита, может оказаться одним из возможных механизмов токсического повреждения печени. Образование нитротирозина в клетках печени также может происходить в присутствии миелопероксидазы [75]. Этот энзим содержится в нейтрофилах, рекрутинг которых при отравлении П наступает на достаточно поздних этапах развития токсической реакции, в связи с чем данный механизм образования и накопления нитротирозина менее вероятен. Известно, что нитротирозин может образовываться и при участии гемма, а также некоторых металлов [76]. Роль этих веществ в образовании нитротирозина при острых отравлениях П не изучена.
Роль цитокинов и других медиаторов воспаления в токсических эффектах, вызываемых парацетамолом.
Сообщения из нескольких исследовательских центров
указывают на вовлеченность провоспалительных цитокинов в
механизмы развития токсического эффекта П. В частности, по
данным Blazka, при отравлении П отмечается повышение концентрации TNF-? и IL-1? [66, 77]. Более того, данными авторами было показано, что иммунная нейтрализация как TNF-?,
так и IL-1? способна на некоторое время снижать выраженность повреждения печени при отравлении П. Однако данные
Simpson et al. [78] не подтвердили терапевтический эффект
блокирования TNF-a при отравлении П.
Наряду с оценкой роли провоспалительных цитокинов в патогенезе токсического действия П были изучены эффекты, связанные с некоторыми противовоспалительными цитокинами. В частности, показано протективное действие IL-10 на клетки печени при введении высоких доз препарата мышам. При отравлениях П отмечено повышение концентрации фактора ингибирования миграции макрофагов (MIF) [68, 79]. Данное вещество обладает многоплановым биологическим действием, объединяя в себе свойства цитокина, гормона и энзима. MIF может вызывать подъем уровня провоспалительных цитокинов, молекул адгезии, экспрессии матрикса металлопротеиназы-2, высвобождения NO и ЦОГ-2, нарушать выработку эндогенных глюкокортикоидов [79]. К каким конкретным патофизиологическим последствиям при отравлении П приводит повышение уровня MIF, в настоящее время не выяснено.
Публикации, касающиеся изменений уровня хемокинов при отравлении П, пока немногочисленны. Тем не менее имеются указания на возможные гепатопротекторные свойства у некоторых представителей этого класса биологически активных веществ [80]. В частности, данное предположение может быть отнесено к протеину, ингибирующему макрофаги-2 (MIP-2). В то же время роль хемоаттрактивного протеина моноцитов-1 (MIP-1) скорее связана с усилением гепатотоксического действия П [81].
Образование супероксида и митохондриальные дисфункции при отравлении парацетамолом. Супероксид может образовываться в ходе ряда реакций, в числе которых участвуют различные ферменты, включая цитохром Р450 [82]. В экспериментальных работах д-ра James по отравлению П была дана оценка метаболической активации (так называемому «респираторному взрыву») клеток Купфера, макрофагов и нейтрофилов [44]. Внезапное резкое повышение утилизации кислорода фагоцитирующими клетками — результат повышения активности фермента НАДФ-оксидазы, однако выполненные исследования показали, что при передозировке П источником повышения уровня супероксида являются не активированные макрофаги. Дальнейшие работы указанных авторов позволили предполагать, что источником образования супероксида служат митохондрии печеночных клеток, под влиянием П находящиеся в состоянии дисфункции. Вероятно, этот механизм играет существенную роль в гепатотоксическом эффекте при передозировке препарата.
Некоторые вопросы лечения острых отравлений парацетамолом
Наиболее эффективным антидотом по отношению к П является N-ацетилцистеин. Это соединение обладает высоким гепатопротекторным эффектом в тех случаях, когда назначается в первые 12 часов после отравления П [36]. При лечении пациентов, поступивших в стационар до истечения первых суток от момента передозировки П, кроме фактора времени следует учитывать уровень препарата в крови. Для облегчения работы практических врачей находят применение несколько вариантов номограмм, при построении которых использованы два параметра — концентрация П и время, прошедшее с момента отравления [17]. Примером такого рода номограмм является таблица Прескотта, в которой отправной точкой служит концентрация П, равная 200 мг/л (через 4 часа с момента отравления), а также концентрация препарата 30 мг/л (через 15 часов с момента отравления) [36]. Сопоставление концентрации П в крови реального больного со значениями номограммы Прескотта позволяет определять степень необходимой интенсивности проводимой терапии и оценить прогноз заболевания в целом. Номограмму целесообразно использовать в первые 15 часов после передозировки П. Большое количество клинических наблюдений позволяет считать, что гепатотоксический эффект при отравлении П возможен при концентрации препарата, равной или превышающей 150 мг/л [17, 83], хотя не все исследователи согласны с этой точкой зрения [42]. Среди отравившихся выделяют по меньшей мере три группы риска, у которых повреждение печени может наступить и при более низкой концентрации П: 1) лица со сниженным уровнем глутатиона в печени (недостаточное питание, нарушения физиологических процессов в кишечнике); 2) состояния, сопровождающиеся повышением активности цитохрома Р450, особенно его изоформ 2Е1, 1А2 и 3А4 (такой эффект может вызывать постоянный прием ряда препаратов, таких как фенитоин и карбамазепин); 3) лица, злоупотребляющие алкогольными напитками [41]. Следует оговориться, что широко распространенное в прошлом представление об особой подверженности алкоголиков тяжелому поражению печени при отравлении сравнительно небольшими дозами П в настоящее время разделяется не всеми специалистами [1].
Лечение N-ацетилцистеином показано в тех случаях, когда в организм больного в течение суток было введено более 150 мг/кг П (для больных из групп повышенного риска — более 75 мг/кг) [17, 83]. К числу наиболее значимых прогностических факторов при отравлении П относят дозу препарата и время, прошедшее от момента приема П до момента госпитализации [41]. Когда госпитализация больного осуществлялась на сроках, превышающих 24 часа, риск возникновения тяжелой печеночной недостаточности существенно увеличивался, что следует связывать с накоплением к концу первых суток в организме пациента метаболитов N-ацетилбензохинонимина и истощением запасов глутатиона в печени [41]. Среди всех больных, госпитализированных поповоду передозировки П (без учета величины дозы препарата и сроков госпитализации), вероятность развития тяжелой печеночной недостаточности составляет около 0,6% [42]. В некоторых странах (США, Великобритания) это обстоятельство позволяет рекомендовать госпитализацию больных с отравлением П в общее терапевтическое отделение и перевод пациента в ОРИТ лишь при появлении лабораторных признаков печеночной недостаточности [41].
Среди наиболее чувствительных прогностических критериев в отношении исхода печеночной недостаточности при отравлении П указывают на величину протромбинового времени. Существует мнемоническое правило, согласно которому увеличение данного показателя > 36 секунд через 36 часов с момента отравления позволяет с высокой вероятностью ожидать развития острой печеночной недостаточности. Напротив, нормализация ранее удлиненного протромбинового времени у больных с развившейся печеночной недостаточностью является хорошим прогностическим признаком [84]. Острая печеночная недостаточность обычно развивается на 4—5-е сутки после отравления. Как правило, тяжелому поражению печени сопутствует синдром полиорганной недостаточности, в который при отравлении П также входят острая почечная недостаточность, гипотония, сепсис, коагулопатии, отек мозга и энцефалопатия [17, 85]. При возникновении тяжелого гепатонекроза следует предусмотреть возможность выполнения операции трансплантации печени по срочным показаниям, так как только это вмешательство способно изменить неблагоприятный прогноз. Целесообразность продолжения терапии N-ацетилцистеином на поздних этапах отравления П остается спорной [83]. Несмотря на отсутствие убедительных теоретических оснований для введения указанного антидота по истечении первых 24-х часов после отравления, в ряде стран и на более поздних сроках N-ацетилцистеин рутинно используют в интенсивной терапии больных, у которых доза принятого внутрь П превысила 150 мг/кг. Так, например, в ОРИТ Англии показанием к отмене N-ацетилцистеина служат нормализация показателей протромбинового времени или выполнение трансплантации печени [17, 86].
Лечение артериальной гипотензии при отравлении П с выраженными явлениями печеночной недостаточности не имеет каких-либо специфических отличий от общепринятых подходов в реанимационной практике [85]. При возникновении явлений острой почечной недостаточности необходимо выполнение гемодиализа [86]. Отек мозга рассматривается как одна из наиболее частых непосредственных причин смерти у данной категории больных. В связи с этим настоятельно рекомендовано проводить динамический контроль уровня внутричерепного давления, особенно у потенциальных реципиентов донорской печени [85, 87]. Безопасным считается уровень дав ления ликвора ниже 200 мм Hg [85].
В одном из сравнительно недавних исследований при оценке тяжести состояния больного и вероятности наступления летального исхода была показана достаточно высокая информативность шкалы APACHE-II [88]. Оценка тяжести состояния больного по шкале APACHE-II может быть полезной и при решении вопроса о целесообразности выполнения пересадки печени. Определенным прогностическим значением обладает уровень лактата крови [86]. Величина данного показателя, превышающая 3,5 ммоль/л в первые 4 часа после госпитализации в центр лечения острых отравлений и > 3,0 ммоль/л после 12 часов от начала интенсивной терапии, является предвестником наступления неблагоприятного исхода.
Заключение
Парацетамол может с успехом использоваться в практической работе ОРИТ. Этот препарат обладает собственным достаточно выраженным обезболивающим и антипиретическим эффектом, а его совместное использование с наркотическими анальгетиками приводит к синергизму действия и снижению расхода опиоидов. При применении в терапевтических дозах парацетамол практически безопасен, но требует взвешенного подхода при назначении пациентам из групп риска, в том числе — алкоголикам.
При передозировке парацетамола органом-мишенью является печень, патогенез повреждения которой сложен и невполне ясен. Тяжелая печеночная недостаточность наиболее часто возникает при сочетании нескольких неблагоприятных факторов: введение высоких доз препарата, несвоевременное и нерациональное оказание интенсивной терапии, особая предрасположенность больного поражениям печени (пациенты из групп риска).
Литература
- Prescott L. F. Paracetamol: past, present and future. Am. J. Ther. 2000; 7: 143—147.
- Davies N. M., Good R. L., Roupe K. A., Yanez J. A. Cyclooxygenase-3: axiom, dogma, anomaly, enigma or splice error? — not as easy as 1, 2, 3. J. Pharm. Pharmaceut. Sci. 2004; 7 (2): 217—226.
- Bannwarth B., Netter P., Lapicque F. et al. Plasma and cerebrospinal fluid concentrations of paracetamol after a single intravenous dose of propac etamol. Br. J. Clin. Pharmacol. 1992; 34: 79—81.
- Piletta P., Porchet H. C., Dayer P. Central analgesic effect of acetaminophen but not of aspirin. Clin. Pharmacol. Ther. 1991; 49: 350—354.
- Romsing J., Moiniche S., Dahl B. Rectal and parenteral paracetamol, and paracetamol in combination with NSAIDs, for postoperative analgesia. Br. J. Anaesth. 2002; 88: 215—226.
- Piquet V., Desmeules J., Dayer P. Lack of acetominophenceiling effect on R-III nociceptive flexion reflux. Eur. J. Clin. Pharmacol. 1998; 53: 321—324.
- Vane J. R. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat. New Bio 1971; 231 (25): 232—235.
- Fu J. Y., Masferrer J. L., Seibert K. et al. The induction and suppression of prostaglandin H2 synthase (cylooxygenase) in human monocytes. J. Biol. Chem. 1990; 265 (28): 1727—1740.
- Simon L. S. COX-2 inhibitors. Are they nonsteroidal anti-inflammatory drugs with a better safety profile? Gastroenterol. Clin. North Am. 2001; 30: 1011—1025.
- Chandrasekharan N. V., Dai H., Roos K. L. et al. COX-3, a cyclooxyge- nase-1 variant inhibited by acetaminophen and other analgesic / antipyretic drugs: cloning, structure, and expression. Proc. Nation. Acad. Sci. USA 2002; 99 (21): 13926—13931.
- Simmons D. L., Botting R. M., Robertson M. et al. Induction of an acetaminophensensitive cyclooxygenase with reduced sensitivity to nons teroid antiinflammatory drugs. Proc. Nation. Acad. Sci. USA. 1999; 96 (6): 3275—3280.
- Zhu X., Conklin D., Eisenach J. C. Cyclooxygenase-1 in the spinal cord plays an important role in postoperative pain. Pain 2003; 104 (1—2): 15—23.
- Graham G. G., Scott K. F. Mechanisms of action of paracetamol and related analgesics. Inflammopharmacology 2003; 11: 401—413.
- Serhan C. N. Lipoxins and aspirin-triggered 15-epilipoxin biosynthesis: an update and role in antiinflammation and pro-resolution. Prostaglandins Other Lipid Mediat. 2002; 68—69: 433—455.
- Serhan C. N., Hong S., Gronert K. et al. Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J. Exp. Med. 2002; 196 (8): 1025—1037.
- Roberts D. W., Bucci T. J., Benson R. W. et al. Immunohistochemical localization and quantification of the 3-(cystein-S-yl)-acetaminophen protein adduct in acetaminophen hepatotoxicity. Am. J. Pathol. 1991; 138: 359—371.
- Wallace C. I., Dargan P. I., Jones A. L.Paracetamol overdose: an evidence based flowchart to guide management. Emerg. Med. J. 2002; 19 (3): 202—205.
- Kehlet H., Dahl J. B. The value of 'multimodal' or 'balanced analgesia' in postoperative pain treatment. Anesth. Analg. 1993; 77: 1048—1056.
- Kehlet H., Dahl J. B. Are perioperative nonsteroidal anti-inflammatory drugs ulcerogenic in the short term? Drugs 1992; 44: 38—41.
- Marret E., Kurdi O., Zufferey P., Bonnet F. Effects of non-steroidal anti-inflammatory drugs on patient-controlled analgesia morphine side effects: meta-analysis of randomized controlled trials. Anesthesiology 2005; 102 (6): 1249—1260.
- Cobby T. F., Crighton I. M., Kyriakides K., Hobbs G. J. Rectal paracetamol has a significant morphine-sparing effect after hysterectomy. Br. J. Anaesth. 1999; 83: 253—256.
- Delbos A., Boccard E. The morphine-sparing effect of propacetamol in orthopedic postoperative pain. J. Pain Symptom Manage 1995; 10: 279—286.
- Peduto V. A., Ballabio M., Stefanini S. Efficacy of propacetamol in the treatment of postoperative pain. Morphine-sparing effect in orthopedic surgery. Italian Collaborative Group on Propacetamol. Acta Anaesthesiol. Scand. 1998; 42: 293—298.
- Remy C., Marret E., Bonnet F. Effects of acetaminophen on morphine side-effects and consumption after major surgery: meta-analysis of randomized controlled trials. Br. J. Anaesthesia 2005; 94 (4): 505—513.
- Benhamou D., Bouaziz H., Zerrouk N., Preaux N. Audit of ketoprofen prescribing after orthopedic and general surgery. Can. J. Anaesth. 1999; 46: 109—113.
- Marret E., Flahault A., Samama C. M., Bonnet F. Effects of postoperative, nonsteroidal, antiinflammatory drugs on bleeding risk after tonsillectomy: meta-analysis of randomized, controlled trials. Anesthesiology 2003; 98: 1497—1502.
- Viel E., Langlade A., Osman M. et al. Propacetamol: from basic action to clinical utilization. Ann. Fr. Anesth. Reanim. 1999; 18: 332—340.
- Aubrun F., Kalfon F., Mottet P. et al. Adjunctive analgesia with intravenous propacetamol does not reduce morphine-related adverse effects. Br. J. Anaesth. 2003; 90: 314—319.
- Fletcher D., Negre I., Barbin С. et al. Postoperative analgesia with i. v. propacetamol and ketoprofen combination after disc surgery. Can. J. Anaesth. 1997; 44: 479—485.
- Hernandez-Palazon J., Tortosa J. A., Martinez-Lage J. F. et al. Intravenous administration of propacetamol reduces morphine consumption after spinal fusion surgery. Anesth. Analg. 2001; 92: 1473—1476.
- Mimoz O., Incagnoli P., Josse C. et al. Analgesic efficacy and safety of nefopam vs. propacetamol following hepatic resection. Anaesthesia. 2001; 56: 520—525.
- Schug S. A., Sidebotham D. A., McGuinnety M. et al. Acetaminophen as an adjunct to morphine by patientcontrolled analgesia in the management of acute postoperative pain. Anesth. Analg. 1998; 87: 368—372.
- Siddik S. M., Aouad M. T., Jalbout M. I. et al. Diclofenac and/or propacetamol for postoperative pain management after cesarean delivery in patients receiving patient controlled analgesia morphine. Reg. Anesth. Pain Med. 2001; 26: 310—315.
- Pettersson P. H., Jakobsson J., Owall A. Intravenous acetaminophen reduced the use of opioids compared with oral administration after coronary artery bypass grafting. J. Cardiothorac. Vasc. Anesth. 2005; 19 (3): 306—309.
- Sinatra R. S., Jahr J. S., Reynolds L. W. et al. Efficacite. antalgique et tole. rance du Perfalgan 1 g dans la douleur postope. ratoire en chirurgie orthopedique. Ann. Fr. Anesth. Reanim. 2001; 20 (Suppl. 1): 170.
- Prescott L. F., Illingworth R. N., Critchley J. A., Proudfoot A. T. Intravenous N-acetylcysteine: the treatment of choice for paracetamol poisoning. B. M. J. 1979; 2: 1097—1100.
- Litovitz T. L., Klein-Schwartz W., Rodgers G. C. et al. 2001 annual report of the American association of poison control centers toxic exposure surveillance system. Am. J. Emerg. Med. 2002; 20: 391—452.
- Litovitz T. L., Klein-Schwartz W., White S. et al. 2000 Annual report of the American association of poison control centers toxic exposure surveillance system. Am. J. Emerg. Med. 2001; 19: 337—395.
- Gyamlani G. G., Parikh C. R. Acetaminophen toxicity: suicidal vs accidental. Crit. Care 2002; 6: 155—159.
- Dargan P. I., Ladhani S. L., Jones A. L. Measuring paracetamol concentrations in all patients with drug overdose or altered consciousness: does it change outcome? Emerg. Med. J. 2001; 18: 178—182.
- Dargan P. I., Jones A. L. Accidental staggered paracetamol overdoses in the UK: epidemiology and outcome. Emerg. Med. J. 2002; 19 (3): 202—205. No abstract available. Erratum in: Emerg. Med. J. 2002; 19 (4): 376.
- Jones A. L., Dargan P. I. Over the counter analgesics: a toxicological perpective. Trends Pharmacol. Sci. 2003; 24 (4): 154—157.
- Bessems J. G., Vermeulen N. P. Paracetamol (acetaminophen)-induced toxicity: molecular and biochemical mechanisms, analogues and protective approaches. Crit. Rev. Toxicol. 2001; 31: 55—138.
- James L. P., McCullough S. S., Knight T. R. et al. Acetaminophen toxicity in mice lacking NADPH oxidase activity: role of peroxynitrite formation and mitochondrial oxidant stress. Free Radic. Res. 2003; 37 (12): 1289—1297.
- James L. P., McCullough S. S., Lamps L. W. et al. Effect of N-acetylcysteine on acetaminophen toxicity in mice: relationship to reactive nitrogen and cytokine formation. Toxicol. Sci. 2003; 75: 458—467.
- Nelson S. G., Wan Z., Stan M. A. S(N)2 ring opening of beta-lactones: an alternative to catalytic asymmetric conjugate additions. J. Org. Chem. 2002; 67: 4680—4683.
- Gujral J. S., Knight T. R., Farhood A. et al. Mode of cell death after acetaminophen overdose in mice: apoptosis or oncotic necrosis? Toxicol. Sci. 2002; 67: 322—328.
- Lawson J. A., Fisher M. A., Simmons C. A. et al. Inhibition of Fas receptor (CD95)-induced caspase activation and apoptosis by acetaminophen in mice. Toxicol. Appl. Pharmacol. 1999; 156: 179—186.
- Mitchell J. R., Jollow D. G., Potter W. Z. et al. Acetaminophen-induced hepatic necrosis. IV. Protective role of glutathione. J. Pharmacol. Exp. Ther. 1973; 187: 211—217.
- Chen W., Koenigs L. L., Thompson S. J. et al. Oxidation of acetaminophen to its toxic quinone imine and nontoxic catechol metabolites by baculovirus-expressed and purified human cytochromes P450 2E1 and 2A6. Chem. Res. Toxicol. 1998; 11: 295—301.
- Patten C. J., Thomas P. E., Guy R. L. et al. Cytochrome P450 enzymesinvolved in acetaminophen activation by rat and human liver microsomes and their kinetics. Chem. Res. Toxicol. 1993; 6: 511—518.
- Thummel K. E., Lee C. A., Kunze K. L. et al. Oxidation of acetaminophen to N-acetyl-p-aminobenzoquinone imine by human CYP3A4. Biochem. Pharmacol. 1993; 45: 1563—1569.
- Pumford N. R., Hinson J. A., Potter D. W. et al. Immunochemical quantitation of 3-(cystein-S-yl)acetaminophen adducts in serum and liver proteins of acetaminophen-treated mice. J. Pharmacol. Exp. Ther. 1989; 248: 190—196.
- Pumford N. R., Roberts D. W., Benson R. W., Hinson J. A. Immunochemical quantitation of 3-(cystein-S-yl)acetaminophen protein adducts in subcellular liver fractions following a hepatotoxic dose of acetaminophen. Biochem. Pharmacol. 1990; 40: 573—579.
- Muldrew K. L., James L. P., Coop L. et al. Determination of acetaminophen-protein adducts in mouse liver and serum and human serum after hepatotoxic doses of acetaminophen using high-performance liquid chromatography with electrochemical detection. Drug Metab. Dispos. 2002; 30: 446—451.
- Nelson S. D. Molecular mechanisms of the hepatotoxicity caused by acetaminophen. Semin. Liver. Dis. 1990; 10: 267—278.
- Tsokos-Kuhn J. O., Hughes H., Smith C. V., Mitchell J. R. Alkylation of the liver plasma membrane and inhibition of the Ca2-ATPase by acetaminophen. Biochem. Pharmacol. 1988; 37: 2125—2131.
- Qiu Y., Benet L. Z., Burlingame A. L. Identification of the hepatic protein targets of reactive metabolites of acetaminophen in vivo in mice using two-dimensional gel electrophoresis and mass spectrometry. J. Biol. Chem. 1998; 273: 17940—17953.
- Coles B., Wilson I., Wardman P. et al. The spontaneous and enzymatic reaction of N-acetyl-p-benzoquinonimine with glutathione: a stopped- flow kinetic study. Arch. Biochem. Biophys. 1988; 264: 253—260.
- Dai Y., Cederbaum A. I. Cytotoxicity of acetaminophen in human cytochrome P4502E1-transfected HepG2 cells. J. Pharmacol. Exp. Ther. 1995; 273: 1497—1505.
- Nakae D., Yoshiji H., Yamamoto K. et al. Influence of timing of administration of liposome-encapsulated superoxide dismutase on its prevention of acetaminophen-induced liver cell necrosis in rats. Acta Pathol. Jpn. 1990; 40: 568—573.
- Sakaida I., Kayano K., Wasaki S. et al. Protection against acetaminophen-induced liver injury in vivo by an iron chelator, deferoxamine. Scand. J. Gastroenterol. 1995; 30: 61—67.
- Schnellmann J. G., Pumford N. R., Kusewitt D. F. et al. Deferoxamine delays the development of the hepatotoxicity of acetaminophen in mice. Toxicol. Lett. 1999; 106: 79—88.
- James L. P., Farrar H. C., Sullivan J. E. et al. Measurement of acetaminophen-protein adducts in children and adolescents with acetaminophen overdoses. J. Clin. Pharmacol. 2001; 41: 846—851.
- Laskin D. L., Gardner C. R., Price V. F., Jollow D. J. Modulation of macrophage functioning abrogates the acute hepatotoxicity of acetaminophen. Hepatology 1995; 21: 1045—1050.
- Blazka M. E., Wilmer J. L., Holladay S. D. et al. Role of proinflammatory cytokines in acetaminophen hepatotoxicity. Toxicol. Appl. Pharmacol. 1995; 133: 43—52.
- Bourdi M., Masubuchi Y., Reilly T. P. et al. Protection against acetaminophen-induced liver injury and lethality by interleukin 10: role of inducible nitric oxide synthase. Hepatology 2002; 35: 289—298.
- Bourdi M., Reilly T. P., Elkahloun A. G. et al. Macrophage migration inhibitory factor in drug-induced liver injury: a role in susceptibility and stress responsiveness. Biochem. Biophys. Res. Commun. 2002; 294: 225—230.
- Hogaboam C. M., Bone-Larson C. L., Steinhauser M. L. et al. Exaggerated hepatic injury due to acetaminophen challenge in mice lacking C-C chemokine receptor 2. Am. J. Pathol. 2000; 156: 1245—1252.
- Goldin R. D., Ratnayaka I. D., Breach C. S. et al. Role of macrophages in acetaminophen (paracetamol)-induced hepatotoxicity. J. Pathol. 1996; 179: 432—435.
- Ju C., Reilly T. P., Bourdi M. Protective role of Kupffer cells in acetaminophen-induced hepatic injury in mice. Chem. Res. Toxicol. 2002; 15 (12): 1504—1513.
- Hinson J. A., Pike S. L., Pumford N. R., Mayeux P. R. Nitrotyrosine-protein adducts in hepatic centrilobular areas following toxic doses of acetaminophen in mice. Chem. Res. Toxicol. 1998; 11: 604—607.
- Pryor W. A., Squadrito G. L. The chemistry of peroxynitrite: a product from the reaction of nitric oxide with superoxide. Am. J. Physiol. 1995; 268 (1): 699—722.
- Beckman J. S., Koppenol W. H. Nitric oxide, superoxide, and peroxynitrite: the good, the bad and ugly. Am. J. Physiol. 1996; 271 (1): С1424—C1437.
- Davis K. L., Martin E., Turko I. V., Murad F. Novel effects of nitric oxide. Annu. Rev. Pharmacol. Toxicol. 2001; 41: 203—236.
- Thomas D. D., Espey M. G., Vitek M. P. et al. Protein nitration is mediated by heme and free metals through Fenton-type chemistry: an alternative to the NO/O2 reaction. Proc. Nation. Acad. Sci. USA 2002; 99: 12691—12696.
- Blazka M. E., Elwell M. R., Holladay S. D. et al. Histopathology of acetaminophen-induced liver changes: role of interleukin 1 alpha and tumor necrosis factor alpha. Toxicol. Pathol. 1996; 24: 181—189.
- Simpson K. J., Lukacs N. W., McGregor A. H. et al. Inhibition of tumor necrosis factor alpha does not prevent experimental paracetamol- induced hepatic necrosis. J. Pathol. 2000; 190; 489—494.
- Baugh J. A., Bucala R. Macrophage migration inhibitory factor. Crit. Care Med. 2002; 30 (Suppl): S27—S35.
- Lawson J. A., Farhood A., Hopper R. D. et al. The hepatic inflammatory response after acetaminophen overdose: role of neutrophils. Toxicol. Sci. 2000; 54: 509—516.
- Hogaboam C. M., Simpson K. J., Chensue S. W. et al. Macrophage inflammatory protein-2 gene therapy attenuates adenovirus- and acetaminophen-mediated hepatic injury. Gene Ther. 1999; 6: 573—584.
- Puntarulo S., Cederbaum A. I. Role of cytochrome P-450 in the stimulation of microsomal production of reactive oxygen species by ferritin. Biochim. Biophys. Acta 1996; 1289: 238—246.
- Jones A. L. Recent advances in the management of late paracetamol poisoning. Emerg. Med. (Aust. ) 2000; 12: 14—21.
- Harrison P. M., O'Grady J. G., Keays R. T. et al. Serial prothrombin time as a prognostic indicator in paracetamol induced fulminant hepatic failure. B. M. J. 1990; 301: 964—966.
- Ellis A., Wendon J. Circulatory, respiratory, cerebral and renal derangements in acute liver failure: pathophysiology and management. Semin. Liv. Dis. 1996; 16: 379—388.
- Bernal W., Donaldson N., Wyncoll D., Wendon J. Blood lactate as an early predictor of outcome in paracetamol-induced acute liver failure: a cohort study. Lancet 2002; 359: 558—563.
- O'Grady J. G., Alexander G. J., Hayllar K. M., Williams R. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology 1989; 97: 439—445.
- Mitchell I., Bihari D., Chang R. et al. Earlier identification of patients at risk from acetaminopehn-induced acute liver failure. Crit. Care Med. 1998; 26: 279—284.